近日,浦合医药在欧洲药物化学杂志 (European Journal of Medicinal Chemistry)公开了其临床3期小分子EGFR抑制剂YK-029A的研发过程。

靶向表皮生长因子受体 (EGFR) 敏感驱动突变del19或L858R (21号外显子),在非小细胞肺癌领域获得了巨大的成功。自第一个EGFR酪氨酸激酶抑制剂(TKI)吉非替尼于2003年被FDA批准后,多个EGFR抑制剂(1代:吉非替尼和厄洛替尼,2代:阿法替尼和达可替尼,3代:奥希替尼)也相继被批准,并表现出了良好的临床疗效。不幸的是,对于EGFR 20外显子插入突变(EGFRex20ins)的非小细胞肺癌患者,其可选择的治疗方式有限,目前主要采用化疗或免疫治疗。此外,敏感突变的患者在使用第一代抑制剂治疗时可能会因获得性T790M耐药而出现疾病复发,而在使用第一代抑制剂的患者中,T790M耐药人群又占据了最高比例。鉴于EGFR突变的非小细胞肺癌患者面临巨大的临床未满足需求,浦合医药希望找到一种新的分子来同时解决上述两个难题。
T790M,也被称为gatekeeper突变,其导致对吉非替尼(或厄洛替尼)的活性降低的原因有如下两点: 1)甲硫氨酸残基侧链与抑制剂发生空间位阻; 2) T790M共突变体(del19/T790M或L858R/T790M)对ATP的亲和力大大增加。T790M共突变也是第二代不可逆TKI (如阿法替尼)的主要耐药机制。最近,奥希替尼被发现可有效抑制T790M共突变患者的肿瘤生长,无进展生存期(PFS)达12.3个月。此外,阿美替尼和氟美替尼也在中国被批准用于治疗T790M共突变的晚期非小细胞肺癌患者。
EGFR20外显子 (氨基酸插入位置 762-774)位于EGFR的C-螺旋处, 大约4-7%的非小细胞患者含有20外显子插入突变,且包含数十种插入类型:包括但不限于A763_Y764insFQEA, V769_D770insASV, D770_N771insSVD, D770_N771insNPG 和 H773_V774ins_NPH等。因此,大部分20外显子插入的结构解析是缺失的。2013年, Yasuda 等人报道了EGFR D770_N771insNPG插入突变的晶体结构,并发现NPG的插入使得EGFR持续激活,但并未降低其与ATP的亲和力。与之相对比的是, FQEA 插入到氨基酸A736_Y764之间,形成了一个额外的螺旋结构, 并使得相邻残基向N端移动,并稳定了L858周围的输水作用,从而使得蛋白处于非活性态。非常不幸的是,由于缺乏足够的安全治疗窗口,基本上所有传统的EGFR抑制剂对于20外显子插入突变均无效。最近, EGFR-MET双抗:amivantamab,被FDA批准用于治疗铂类耐药后的2线EGFRex20ins插入患者;mobocertinib (TAK-788; Figure 1) 也基于其治疗2线EGFRex20ins的出色 ORR (43%) 被FDA批准。尽管如此,目前在EGFRex20ins初治患者(1线)中,尚未有良好的治疗手段。浦合医药的新一代EGFR抑制剂YK-029A目前正在开展头对头含铂化疗在1线EGFR20外显子插入初治患者中的三期临床试验。
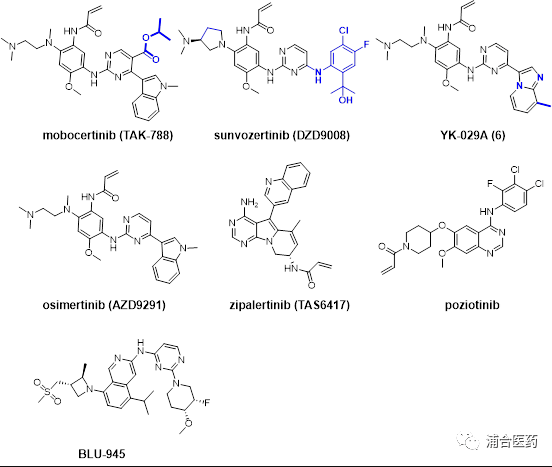
Figure 1. 代表性EGFR抑制剂结构
靶向EGFRex20ins设计的最大挑战就是对于野生型EGFR (wt-EGFR)的选择性。敏感突变 (del19 或L858R) 相比 wt-EGFR对于ATP的结合力大大降低;而 EGFRex20ins突变则提高了与ATP的亲和力。因此,对于20号外显子插入突变活性的提高通常也使得选择性降低,而失去治疗窗口。脱靶野生型EGFR的抑制在临床上会带来一些副作用,例如皮疹和腹泻。使用160 mg mobocertinib最常见的副作用包括(TRAEs; 30%):腹泻 (90%), 皮疹 (45%)以及甲沟炎(34%), 并有17%停药率。
在临床中发现, 一些EGFRex20ins患者可以通过off-lable使用更高剂量的奥西替尼 (160 mg)来展现一些临床效果(奥希替尼推荐剂量80 mg),但同时带来了更多的毒副作用。因此,我们意识到,如果提高其对于20外显子插入突变的活性和选择性,可能带来更优的临床治疗效果。Phe723是一个在P-loop的保守氨基酸,不论是T790M,还是wt (Figure 2),甚至是20外显子插入,其均在相同的位置上,因此我们希望提高对于EGFR (mutant or wt) reversible binding affinity,来达到最终活性的提高。由于EGFRex20ins在激酶结构域ATP结合区的氨基酸序列与野生型完全一致,因此我们可以用野生型EGFR来模拟EGFRex20ins (Figure 2)。此外,由于 wt-EGFR与ATP的结合力最强 [Ki = 5 μM], 通过增加与EGFR可逆的结合力,最终也会增加EGFRex20ins的活性,由于ATP结合力的不同,从而使得分子能增加活性的同时保持选择性。
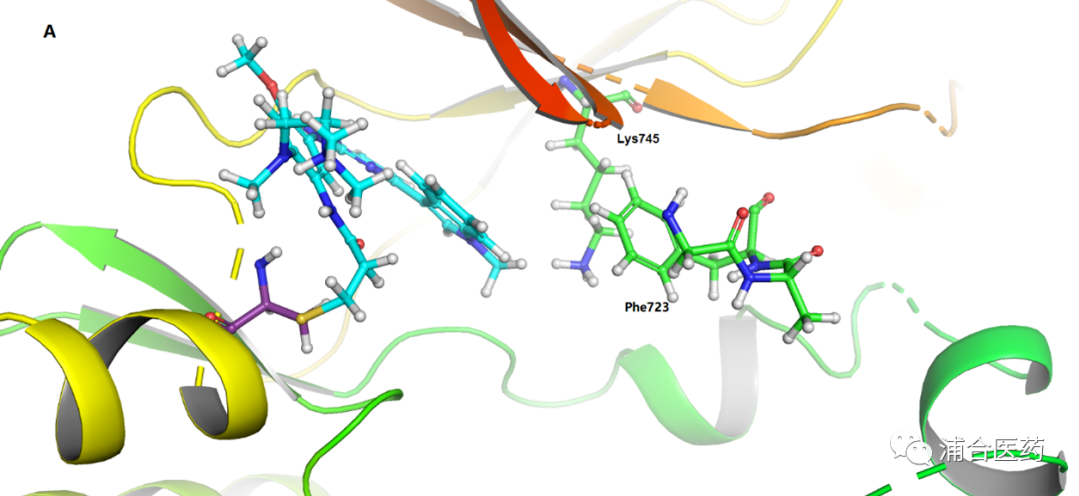
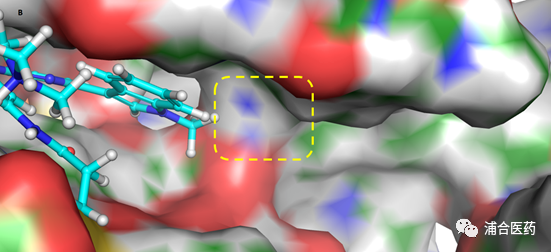
Figure 2A. The X-ray crystal structure of osimertinib bound to the wt-EGFR with 2.31 Å resolution (PDB:6JXT), osimertinib (cyan), Phe723 and Lys745 of EGFR (green); 2B. Surface view (yellow box: hydrophobic pocket near Phe723).
经过多轮的优化筛选,最终找到了候选分子YK-029A。其对于T790M的结合与EGFRex20ins的结合模式类似 (Figure S1),并与设计理念吻合,通过与Phe723的疏水作用 (Figure 3),达到了活性的提高和选择性的保持。此外,还有一个不稳定(推测可被水分子介导)的和Lys745的氢键作用。
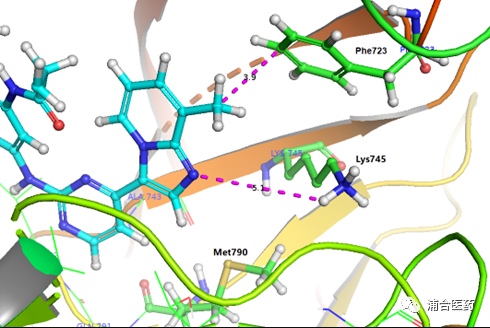
Figure 3. Predicted binding mode of YK-029Ain EGFRLR/TM (PDB:6JX0). Protein: ribbon and green (Phe723, Lys745 and Met790); YK-029A: cyan.
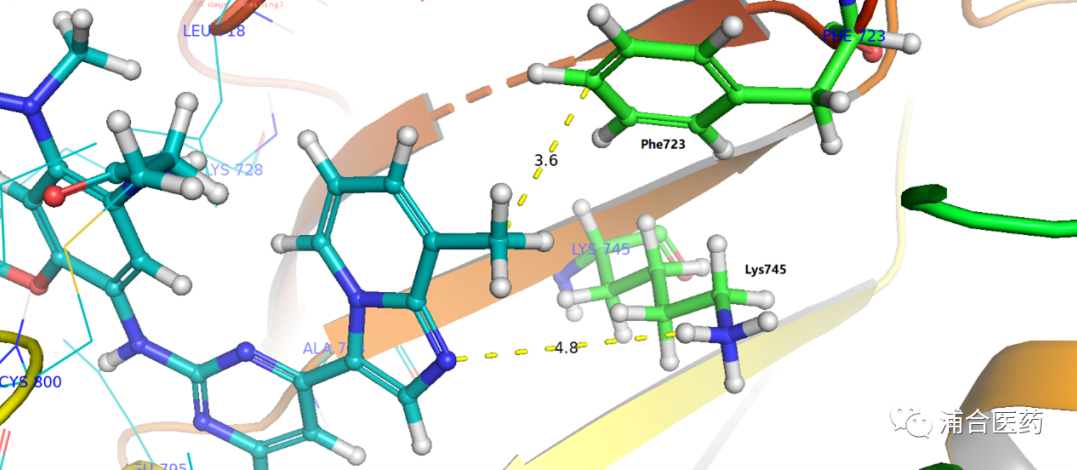
Figure S1. Predicted binding mode of YK-029A in EGFRex20ins (insNPG, PDB:7LGS).
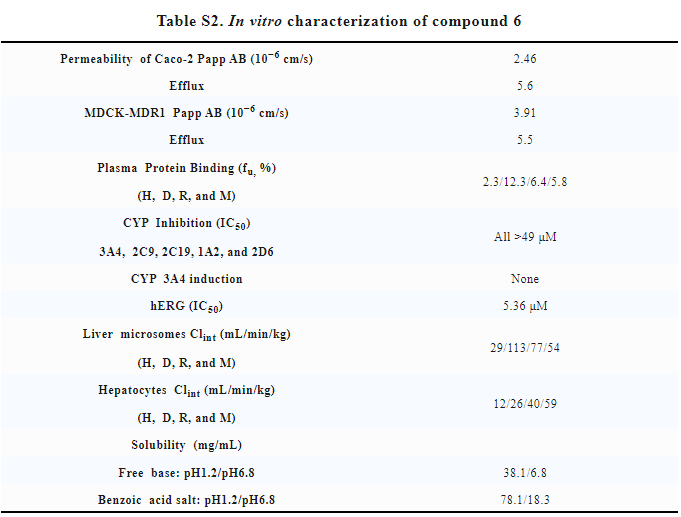
此外,YK-029A的体外性质良好(Table S2),对于多个CYP亚型没有明显的抑制作用,也无诱导效果。人体肝细胞代谢稳定,溶解度和透膜性良好,具有不错的口服生物利用度。
药效方面,YK-029A不论是在T790M共突变的CDX模型还是20外显子插入的PDX模型均展现了良好的体内抑制效果 (Figure 5A、5B)。高剂量组均可使得肿瘤消退,并没有明显的毒副作用,小鼠体重降低控制在5%以内。此外,在PK-PD模型中,也再次验证了YK-029A的靶向作用(Figure 6A、6B)。
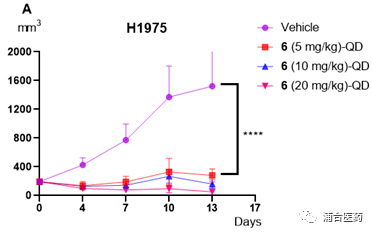
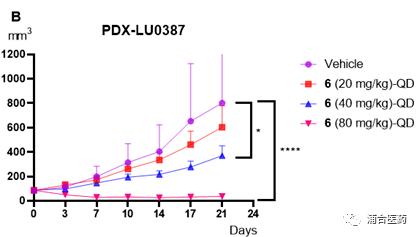
Figure 5A. Antitumor efficacy of compound 6 in the HCC1975 xenograft model of female BALB/c mice (n = 8/group); 5B. Effects of compound 6 in the LU0387 human NSCLC PDX model of BALB/c nude mice (n = 7/group); *p<0.05; **** p<0.0001.
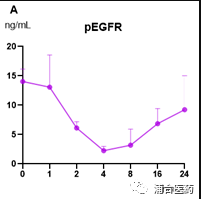
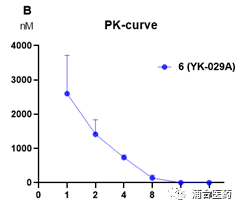
Figure 6A. The pEGFR signal in the LU0387 model after oral administration of compound 6 (80 mg/kg); 6B. The PK curve of compound 6 in the BALB/c nude mice.
在大鼠28天的GLP毒理实验中, 高剂量YK-029A (80 mg/kg)展现了良好的安全治疗窗口,与之相对比的是奥希替尼(80 mg/kg, 大部分大鼠死亡)。在临床剂量爬坡实验中,200 mg剂量下的YK-029A没有明显的严重副作用 [low grade diarrhea (≥Grade 3: 15%) and rash (≥Grade 3: 0%)],与临床前观察到的安全性保持一致。
结论:通过计算机辅助药物设计, YK-029A 被确定为新一代的EGFR抑制剂,可以同时克服敏感突变T790M耐药,以及EGFR20外显子插入突变。由于缺乏精确的晶体结构,通过奥希替尼和野生型EGFR的晶体蛋白结构分析:发现了一个未被其占据的疏水口袋。浦合医药利用该口袋设计出了新一代不可逆共价抑制剂YK-029A,YK-029A在小鼠体内 EGFRLR/TMCDX 和EGFRex20insPDX模型均展现了良好的肿瘤抑制效果。这些数据支持 YK-029A其向临床开展,以期使得更多的非小细胞肺癌患者获益。目前YK-029A的三期临床试验正在进行中。

